| 2.3.5
Angeborene Herzfehler |
|
_____ |
| Von
1000 lebend geborenen Kindern weisen 8-10 einen angeborenen Herzfehler
auf. Lebenserwartung und -qualität können bei ca. 90% der betroffenen
Kinder vor allem durch herzchirurgische Maßnahmen deutlich verbessert
werden. |
|
Häufigkeit |
|
Tab. 14: Häufigkeitsverteilung
wichtiger angeborener Herzfehler (Deutsches Herzzentrum München) nach
G. Rieker
| 1. Linksobstruktion |
| Aortenstenose |
| Aortenisthmusstenose |
|
|
| 2. Rechtsobstruktion |
| Pulmonalstenose |
| Fallottetralogie |
| Pulmonalatresie |
| Trikuspidalatresie |
| Ebsteinanomalie |
|
| - |
| 10,6% |
| 5,3% |
| 1,0% |
| 1,0% |
| 0,5% |
|
| 3. Septumdefekte |
| Ventrikelseptumdefekt |
| Offener Ductus
arteriosus Botalli |
| Vorhofseptumdefekt |
| AV-Septumdefekt |
|
|
| 4. Fehlabgänge
großer Arterien |
| Komplette
Transposition |
|
|
|
|
|
Tab.
14 |
|
|
| 2.3.5.1
Allgemeines |
|
| Der menschliche Fetus besitzt
in den ersten Lebenswochen nur einen Vorhof, eine Herzkammer und einen
herznahen Arterienstamm. Die Ausbildung von zwei Herzteilen und zwei Kreisläufen
ist nur möglich, weil Scheidewände (Septen) die Herzhöhlen
und die großen Gefäße unterteilen. Dazu kommen komplizierte
Drehungsvorgänge. Störungen dieser fetalen Herzentwicklung sind
die Ursache der angeborenen Herzfehler. |
|
Entstehung |
Drei Grundstörungen
können auftreten:
| |
Es bestehen abnorme Verbindungen
zwischen kleinem und großem Kreislauf (z.B. Vorhof- oder Kammerseptumdefekt,
offener Ductus Botalli). |
| |
Es entstehen Stenosen
(z.B. Pulmonalstenose, Aortenisthmusstenose). |
| |
Es liegt eine Transposition,
d.h. eine Verlagerung der großen Gefäße vor (z.B. Abgang
der Aorta aus der rechten, der Arteria pulmonalis aus der linken Herzkammer). |
|
| Besteht auf Grund einer
solchen Fehlentwicklung eine Kurzschlussverbindung zwischen einzelnen Herz-
oder Gefäßabschnitten, so spricht man von einem Shunt.
Beim sogenannten Rechts-Links-Shunt wird venöses dem arteriellen,
beim Links-Rechts-Shunt arterielles dem venösen Blut beigemischt.
Die Prognose der Herzfehler mit Rechts-Links-Shunt ist im allgemeinen
wesentlich ungünstiger. Nicht selten treten angeborene Herzfehler
im Rahmen anderer Missbildungen auf, z.B. beim Down-Syndrom. |
Als Ursachen der genannten
Entwicklungssstörungen kommen neben genetischen Faktoren vor allem
schädigende Einflüsse während der Frühschwangerschaft
(1. und 2. Schwangerschaftsmonat) in Frage. Am besten erforscht ist das
sogenannte Gregg-Syndrom: Je nachdem, ob die Mutter im ersten, zweiten
oder dritten Schwangerschaftsmonat an Röteln erkrankt, kommt
es zu Augenmissbildungen, Herzmissbildungen bzw. Innenohrschäden.
Manche Herzfehler, besonders
diejenigen mit großem Rechts-Links-Shunt, gehen bereits nach der
Geburt mit einer schweren Zyanose einher, weil venöses Blut dem arteriellen
beigemischt wird (sog. blue babies). Es besteht also eine Frühzyanose.
Andere angeborene Herzfehler führen erst zu einem späteren Zeitpunkt
im Leben zu einer meist auch schwächeren Zyanose (Spätzyanose).
Schließlich gibt es eine Gruppe von angeborenen Herzfehlern, die
zu keinem Zeitpunkt eine nennenswerte Zyanose entwickeln. Diese Einteilung
in eine frühzyanotische und spätzyanotische Gruppe sowie
in die Gruppe ohne Zyanose ist von praktischer Bedeutung. |
|
Ursachen |
|
|
Abb.
16: Angeborene Herzfehler
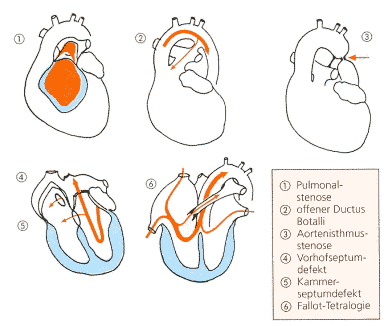 |
|
Abb.
16 |
|
|
| Die Zyanose ist ein wichtiges,
aber keineswegs regelmäßiges oder von Anfang an bestehendes
Symptom. Eine Polyglobulie sowie Trommelschlägelfinger und
-zehen finden sich praktisch nur in der Gruppe der sog. blue babies.
Die Kinder sind nicht selten unterentwickelt. Häufig besteht eine
Neigung zu Bronchitiden. Typisch ist die Hockerstellung, das sog. Squatting
der Kinder mit Fallotscher Tetralogie. Eine schwerwiegende, relativ häufige
Komplikation stellt die Disposition für eine bakterielle Endokarditis
an den missgebildeten Herz- und Gefäßabschnitten dar (s. Abb.
16). |
|
Klinisches
Bild |
| Die meisten angeborenen
Herzfehler müssen am eröffneten, blutleeren Herzen operativ angegangen
werden (sog. offene Operationsmethode); Kreislauf und Atmung werden während
dieser Phase von der Herz-Lungen-Maschine übernommen (sog. extrakorporaler
Kreislauf). |
|
Therapie |
|
|
| 2.3.5.2
Angeborene Herzfehler ohne Shunt |
|
| Definition: Die Aorta
zeigt an der Stelle der früheren Einmündung des sog. Ductus Botalli,
der in der Fetalzeit Arteria pulmonalis und Aorta verbindet, nach der Geburt
jedoch verödet, eine hochgradige Einengung. |
|
|
|
 |
|
Als Folge der Aortenisthmusstenose
herrscht in den arteriellen Kreislaufabschnitten vor der Stenose ein sehr
hoher, dahinter ein sehr niedriger Blutdruck. Die Patienten haben als Leitsymptom
einen Bluthochdruck im Bereich der Arme und einen erniedrigten
Blutdruck an den unteren Extremitäten.
Die Diagnose kann manchmal
mit "zwei Fingern" gestellt werden: während der Arteria-radialis-Puls
sehr kräftig schlägt, ist der Arteria-femoralis-Puls am Leistenband
nur sehr schwach tastbar oder fehlt vollständig. Eine Diagnosesicherung
ist durch Computer- und Kernspintomographie möglich.
Als Komplikation kann eine
bakterielle Endokarditis hinzutreten. Todesursache ist die Linksherzinsuffizienz.
Die Behandlungsmethode der Wahl ist die Operation. |
|
Aortenisthmusstenose
(Erwachsenenform) |
|
| Definition: Bei der
Pulmonalstenose handelt es sich in 90% der Fälle um eine Stenose der
Pulmonalklappe, d.h. um eine valvuläre Stenose. Seltener liegt eine
Stenosierung in dem darunter gelegenen Gefäßabschnitt, dem Infundibulum,
vor (infundibuläre Stenose). |
|
|
|
|
|
| Leitsymptom ist die in der
Jugend zunehmende Atemnot, Todesursache das Rechtsherzversagen. Bei einem
Druckgradienten über 50 mm Hg ist heute die perkutane Ballondilatation
die Methode der Wahl. |
|
Pulmonalstenose |
|
|
| nach oben |
 |
|
|
|
|
| 2.3.5.3 Angeborene Herzfehler
mit Links-Rechts-Shunt |
| Vorhofseptumdefekt (ASD) |
|
| Definition: Die Vorhofscheidewand
weist einen Defekt mit Links-Rechts-Shunt auf. |
|
|
|
|
|
Der Vorhofseptumdefekt ist
der häufigste angeborene Herzfehler. In der Kindheit und Jugend
besteht eine Neigung zu Bronchitis und Lungenentzündung, später
treten Atemnot und Zyanose hinzu. Eine aufgepfropfte bakterielle Endokarditis
kann das Krankheitsbild komplizieren. Häufig besteht eine Kombination
mit anderen Missbildungen (s. Fallotsche Erkrankung, S. 114  ).
Schließlich führt ein Rechtsherzversagen zum Tode. Bei großen
Defekten mit einem Shuntvolumen von > 50 % ist eine möglichst frühzeitige
Operation anzustreben. ).
Schließlich führt ein Rechtsherzversagen zum Tode. Bei großen
Defekten mit einem Shuntvolumen von > 50 % ist eine möglichst frühzeitige
Operation anzustreben. |
|
Vorhofseptumdefekt
(ASD) |
|
|
| Kammerseptumdefekt (VSD) |
|
| Definition: In der
Kammerscheidewand findet sich ein Defekt, der zwischen 0,5 bis 5 cm2
groß sein kann. |
|
|
|
|
|
| Es kommt nicht nur isoliert,
sondern häufig kombiniert vor und ist daher bei 25% aller angeborenen
Herzfehler nachweisbar. Auch hier findet sich eine Neigung zu Infekten
der oberen Luftwege. Eine Zyanose tritt erst in den Spätstadien auf.
Links- und Rechtsherzversagen führen schließlich (bei schweren
Fällen bereits im ersten Lebensjahr) zum Tode. Operiert werden sollte
bei mittlerem bis großem Defekt so früh wie möglich nach
dem 3. Lebensjahr. Beim sog. Morbus Roger, d.h. einem kleinen Ventrikelseptumdefekt,
ist eine Operation nicht erforderlich. |
|
Kammerseptumdefekt
(VSD) |
|
|
| Offener Ductus botalli |
|
| Definition: Offenbleiben
des Ductus Botalli, der während der Fetalzeit Arteria pulmonalis und
Aorta verbindet. |
|
|
|
|
|
| Der offene Ductus Botalli
kann eine sehr unterschiedliche Größe aufweisen. Es strömt
Blut aus der Aorta ständig über die Arteria pulmonalis in den
kleinen Kreislauf zurück (Links-Rechts-Shunt). Die Blutdruckamplitude
wird dadurch, wie bei der Aorteninsuffizienz, sehr groß, der diastolische
Blutdruck niedrig. In einem Drittel der Fälle besteht zunächst
eine bakterielle Endokarditis. Die Operation, die in einer doppelten Unterbindung
des Ductus Botalli besteht, sollte immer angestrebt werden. |
|
Offener
Ductus botalli |
|
|
| 2.3.5.4
Angeborene Herzfehler mit Rechts-Links-Shunt |
|
| Leitsymptom ist die
von Anfang an bestehende, ständig zunehmende Zyanose. Später
tritt Atemnot hinzu, häufiger pfropft sich eine Endokarditis auf.
Die wichtigsten angeborenen Herzfehler mit Rechts-Links-Shunt wurden 1888
von dem französischen Arzt FALLOT ausführlich beschrieben, weshalb
sie unter dem Sammelbegriff Fallot-Syndrom zusammengefaßt werden.
Je nachdem, ob sich 3, 4 oder 5 Fehler kombinieren, spricht man von Fallotscher
Trilogie, Tetralogie bzw. Pentalogie. Immer besteht eine infundibuläre
Pulmonalstenose und eine Rechtsherzhypertrophie. Diese Fehler können
kombiniert sein mit einem Vorhofseptumdefekt, einem Kammerseptumdefekt
oder einer Aorta, die nach rechts verlagert ist, über dem Kammerseptum
"reitet" (reitende Aorta) und daher Blut aus beiden Kammern bekommt (s.
Abb. 16 auf S. 111 ). |
|
Leitsymptome |
| Fallotsche
Trilogie |
= Pulmonalstenose + Rechtshypertrophie
+ Vorhofseptumdefekt |
| Fallotsche
Tetralogie |
= Pulmonalstenose + Rechtshypertrophie
+ reitende Aorta + Kammerseptumdefekt |
| Fallotsche
Pentalogie |
= Fallotsche Tetralogie
+ Vorhofseptumdefekt |
|
|
Einteilung |
| Die Lebenserwartung der
Patienten ist erheblich reduziert, mit 20 Jahren sind nur noch 3-5% der
Patienten ohne operative Korrektur am Leben. |
|
Prognose |
|
|
| 2.3.6
Kardiomyopathien |
|
| Definition: Kardiomyopathien
sind relativ seltene Herzmuskelerkrankungen, die nicht auf eine der üblichen
Ursachen wie beispielsweise Hypertonie, koronare Herzkrankheit oder Herzklappenfehler
zurückzuführen sind. |
|
|
|
|
|
| Bei der hypertrophen obstruktiven
Kardiomyopathie (HOCM) liegt eine starke Hypertrophie der Kammermuskulatur
des linken Ventrikels vor, so daß während der Systole der Auswurf
des Blutes aus der Kammer behindert wird. Wahrscheinlich handelt es sich
um eine vererbbare Erkrankung. Digitalis führt zu einer Verschlechterung
der Symptome und ist daher kontraindiziert. Eine Besserung kann mit Beta-Rezeptorenblockern
oder Calciumantagonisten (z.B. Isoptin®) erreicht werden. Gelegentlich
kommt als großer operativer Eingriff die Entfernung der hypertrophierten
Myokardanteile in Frage. |
|
HOCM |
| Bei der hypertrophen nicht
obstruktiven Kardiomyopathie (HNOCM) ist der Blutauswurf aus der linken
Kammer nicht behindert. |
|
HNOCM |
Die kongestive oder auch
dilatative Kardiomyopathie (CCM) ist durch eine erhebliche Erweiterung
des Ventrikels gekennzeichnet. Sie kann unterschiedliche Ursachen
haben:
unerkannte
Virusmyokarditis,
chronischer
Alkoholkonsum (Alkohol-Kardiomyopathie),
Kollagenosen,
Medikamente
(z.B. Zytostatika wie Adriamycin, Cyclophosphamid u.a.). |
|
CCM |
| Zur Behandlung werden dieselben
therapeutischen Maßnahmen wie bei der Therapie der Herzinsuffizienz
eingesetzt. |
|
Therapie |
|
|
| nach oben |
|
|
|
|
|
| 2.3.7 Cor pulmonale |
|
| Definition: Hypertrophie
oder Dilatation des rechten Herzens durch eine mit pulmonaler Hypertonie
einhergehende Erkrankung. |
|
|
|
|
|
| Das Cor pulmonale stellt
daher eine spezielle Form der Überlastung des rechten Herzens
dar. Wie der Name besagt (lat. pulmo = Lunge), spielt bei seiner Entstehung
die Lunge eine wesentliche Rolle. |
| Wird aus irgendeinem Grund
die Lungenstrombahn, d.h. der kleine Kreislauf, stärker eingeengt,
so muß das rechte Herz das Blut gegen einen höheren Widerstand
durch die Lungen treiben, was zu einem Druckanstieg im kleinen Kreislauf,
d.h. zur pulmonalen Hypertonie führt. Diese Mehrbelastung löst
zunächst eine Hypertrophie, später eine Dilatation des rechten
Ventrikels aus. Ist die Einengung der Lungenstrombahn hochgradig oder besteht
sie über lange Zeit, so kommt es schließlich zum Versagen, d.h.
zur Insuffizienz des rechten Herzens. |
|
Entstehung |
| Werden plötzlich
größere Lungengefäßabschnitte durch Thromben aus
dem venösen Teil des großen Kreislaufs - z.B. aus den Beinvenen
- verlegt, liegt also eine Lungenembolie vor, so entwickelt sich
innerhalb kurzer Zeit eine akute Rechtsherzüberlastung, das akute
Cor pulmonale. |
|
Ursachen |
| Wird die Lungenstrombahn
allmählich
durch eine chronische Lungenerkrankung wie z.B. ein Lungenemphysem,
eine Lungenfibrose oder durch chronisch-rezidivierende Lungenembolien eingeengt,
so kann im Laufe der Jahre ein chronisches Cor pulmonale entstehen. Häufigste
Ursache eines chronischen Cor pulmonale sind die obstruktiven Atemwegserkrankungen. |
| Besteht aus unbekannten
Gründen ein Hochdruck im Lungenkreislauf, so liegt eine primäre
pulmonale Hypertonie vor. Diese sehr seltene Erkrankung entsteht möglicherweise
auf der Grundlage einer angeborenen Strukturveränderung der Lungengefäße. |
| Das klinische Bild wird
von der zugrunde liegenden Lungenerkrankung mitbestimmt. Beim chronischen
Cor pulmonale besteht häufig eine ausgeprägte Zyanose, weil zum
einen der Gasaustausch (Sauerstoff) in der erkrankten Lunge gestört
ist und zum anderen eine Insuffizienz des rechten Herzens vorliegt. Meist
bestehen zudem eine Dyspnoe, Trommelschlägelfinger und -zehen
sowie eine Polyglobulie durch chronischen Sauerstoffmangel. Die
Symptome des akuten Cor pulmonale werden bei der Lungenembolie geschildert
(s. Kap. 5.4.9). |
|
Klinisches
Bild |
Das insuffiziente chronische
Cor pulmonale wird wie jede Herzinsuffizienz mit Digitalis und Diuretika
behandelt. Vor allem muss aber die broncho-pulmonale Grunderkrankung intensiv
angegangen werden. Bei chronisch-obstruktiven Atemwegserkrankungen wirkt
die Sauerstoff-Langzeittherapie nachweislich lebensverlängernd.
Das Atemzentrum spricht
bei vielen dieser Patienten nur noch auf den Sauerstoffmangelreiz
an. Wird zuviel Sauerstoff gegeben, so entfällt der einzige
Reiz für das Atemzentrum, und es entwickelt sich ebenfalls rasch eine
Atemlähmung.
Ebenso ist es gegen Opiate und Sedativa überempfindlich. |
|
Therapie |
|
| Merke: Patienten
mit chronischem Cor pulmonale dürfen ohne ausdrückliche ärztliche
Anweisung weder Morphin, Morphinderivate, Schlaf- oder Beruhigungsmittel
noch Sauerstoff gegeben werden. |
|
|
|
 |
|
| Die Prognose ist recht ungünstig,
da die Ursache der Rechtsherzüberlastung meist nicht beseitigt werden
kann. Zwei Jahre nach den ersten Insuffizienzzeichen des chronischen Cor
pulmonale lebt nur noch knapp die Hälfte der Patienten. |
|
Prognose |